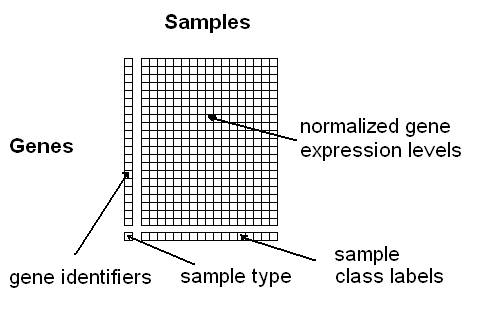
ArrayMining - Online Microarray Data Mining
Ensemble and Consensus Analysis Methods for Gene Expression Data
 Contact Contact |
[X] |
|
The project and the website are maintained by Enrico Glaab School of Computer Science, Nottingham University, UK  webmaster@arraymining.net webmaster@arraymining.net
|
|
| Close | |
|
Hall's CFS - Combinatorial Feature Selection |
[X] |
|
Short description: Hall's CFS is a combinatorial correlation-based feature selection algorithm. A greedy-best first search strategy is used to identify features with high correlation to the response variable but low correlation amongst each other based on the following scoring function: 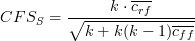
where S is the selected subset with k features and 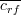 is the average
feature-class correlation and is the average
feature-class correlation and 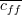 the average feature-feature correlation. the average feature-feature correlation.
References: - Hall, M.A., Correlation-based feature selection for discrete and numeric class machine learning, Proceedings of the Seventeenth International Conference on Machine Learning (2000), p. 359-366 |
|
|
PLS-CV - Partial Least Squares Cross-Validation |
[X] |
|
Short description: The importance of features is estimated based on the magnitudes of the coefficients obtained from training a Partial Least Squares classifier. The number of PLS-components n is selected based on the cross-validation accuracies for 20 random 2/3-partitions of the data for all possible values of n. We use the PLS-implementation in R by Boulesteix et al. References: - Hall, M.A., Correlation-based feature selection for discrete and numeric class machine learning, Proceedings of the Seventeenth International Conference on Machine Learning (2000), p. 359-366 |
|
|
Significance analysis of microarrays (SAM) |
[X] |
|
Short description: SAM (Tusher et al., 2001) is a method to detect differentially expressed genes that uses permutations of the measurements to assign significance values to selected genes. Based on the expression level change in relation to the standard deviation across the measurements a score is calculated for each gene and the genes are filtered according to a user-adjustable threshold (delta). The False Discovery Rate (FDR), i.e. the percentage of genes selected by chance, is then estimated from multiple permutations the measurements. We use the standard SAM-implementation from the samr-package (v1.25). References: - Tusher, V., Tibshirani, R. and Chu, G.: Significance analysis of microarrays applied to the ionizing radiation response", PNAS 2001 (98), p. 5116-5121 |
|
|
Empirical Bayes moderated t-test (eBayes) |
[X] |
|
Short description: The empirical Bayes moderated t-statistic (eBayes, Loennstedt et al., 2002) ranks genes by testing whether all pairwise contrasts between different outcome-classes are zero. An empirical Bayes method is used to shrink the probe-wise sample-variances towards a common value and to augment the degrees of freedom for the individual variances (Smyth, 2004). For multiclass problems the F-statistic is computed as an overall test from the t-statistics for every genetic probe. We use the eBayes-implementation in the R-package limma (v2.12). References: - Loennstedt, I. and Speed, T. P. (2002). Replicated microarray data. Statistica Sinica 12, 31-46. - Smyth, G. K. (2004). Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology, 3, No. 1, Article 3 |
|
|
RF-MDA - Random Forest Feature Selection |
[X] |
|
Short description: A random forest (RF) classifier with 200 trees is applied and the importance of features is estimated by means of the mean decrease in accuracy (MDA) for the out-of-bag samples. We use the RF implementation from the "randomForest" R-package based on L. Breiman's random forest algorithm. References: - Breiman, L. (2001), Random Forests, Machine Learning 45(1), p. 5-32 |
|
|
ENSEMBLE - Ensemble Feature Selection |
[X] |
|
Short description: This selection method combines three univariate filters to an ensemble feature ranking. The used filters are a correlation filter based on the absolute Pearson correlation between a feature vector and the outcome vector, a signal-to-noise-ratio (SNR) filter which extends the SNR-measure to multiple classes based on the pairwise SNRs, and an F-score filter. All filters receive the same weight and the final ranking is obtained as the sum of the individual ranks. |
|
|
Golub et al. (1999) Leukemia data set |
[X] |
|
Short description: Analysis of patients with acute lymphoblastic leukemia (ALL, 1) or acute myeloid leukemia (AML, 0). Sample types: ALL, AML No. of genes: 7129 No. of samples: 72 (class 0: 25, class 1: 47) Normalization: VSN (Huber et al., 2002) References: - Golub et al., Molecular Classification of Cancer: Class Discovery and Class Prediction by Gene Expression Monitoring, Science (1999), 531-537 - Huber et al., Variance stabilization applied to microarray data calibration and to the quantification of differential expression, Bioinformatics (2002) 18 Suppl.1 96-104 |
|
|
van't Veer et al. (2002) Breast cancer data set |
[X] |
|
Short description: Samples from Breast cancer patients were subdivided in a "good prognosis" (0) and "poor prognosis" (1) group depending on the occurrence of distant metastases within 5 years. The data set is pre-processed as described in the original paper and was obtained from the R package "DENMARKLAB" (Fridlyand and Yang, 2004). Sample types: good prognosis, poor prognosis No. of genes: 4348 (pre-processed) No. of samples: 97 (class 0: 51, class 1: 46) Normalization: see reference (van't Veer et al., 2002) References: - van't Veer et al., Gene expression profiling predicts clinical outcome of breast cancer, Nature (2002), 415, p. 530-536 - Fridlyand,J. and Yang,J.Y.H. (2004) Advanced microarray data analysis: class discovery and class prediction (https://genome. cbs.dtu.dk/courses/norfa2004/Extras/DENMARKLAB.zip) |
|
|
Yeoh et al. (2002) Leukemia multi-class data set |
[X] | ||
|
Short description: A multi-class data set for the prediction of the disease subtype in pediatric acute lymphoblastic leukemia (ALL).
No. of samples: 327 Normalization: VSN (Huber et al., 2002) References: - Yeoh et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. March 2002. 1: 133-143 - Huber et al., Variance stabilization applied to microarray data calibration and to the quantification of differential expression, Bioinformatics (2002) 18 Suppl.1 96-104 |
|||
|
Alon et al. (1999) Colon cancer data set |
[X] |
|
Short description: Analysis of colon cancer tissues (1) and normal colon tissues (0). Sample types: tumour, healthy No. of genes: 2000 No. of samples: 62 (class 1: 40, class 0: 22) Normalization: VSN (Huber et al., 2002) References: - U. Alon, N. Barkai, D. Notterman, K. Gish, S. Ybarra, D. Mack, and A. Levine, �Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays,� in Proceedings of the National Academy of Science (1999), vol. 96, pp. 6745�6750 - Huber et al., Variance stabilization applied to microarray data calibration and to the quantification of differential expression, Bioinformatics (2002) 18 Suppl.1 96-104 |
|
|
Singh et al. (2002) Prostate cancer data set |
[X] |
|
Short description: Analysis of prostate cancer tissues (1) and normal tissues (0). Sample types: tumour, healthy No. of genes: 2135 (pre-processed) No. of samples: 102 (class 1: 52, class 0: 50) Normalization: GeneChip RMA (GCRMA) References: - D. Singh, P.G. Febbo, K. Ross, D.G. Jackson, J.Manola, C. Ladd, P. Tamayo, A.A. Renshaw, A.V. D�Amico, J.P. Richie, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell, 1(2): pp. 203�209, 2002 - Z. Wu and R.A. Irizarry. Stochastic Models Inspired by Hybridization Theory for Short Oligonucleotide Arrays. Journal of Computational Biology, 12(6): pp. 882�893, 2005 |
|
|
Shipp et al. (2002) B-Cell Lymphoma data set |
[X] |
|
Short description: Analysis of Diffuse Large B-Cell lymphoma samples (1) and follicular B-Cell lymphoma samples (0). Sample types: DLBCL, follicular No. of genes: 2647 (pre-processed) No. of samples: 77 (class 1: 58, class 0: 19) Normalization: VSN (Huber et al., 2002) References: - M.A. Shipp, K.N. Ross, P. Tamayo, A.P. Weng, J.L. Kutok, R.C.T. Aguiar, M. Gaasenbeek, M. Angelo, M. Reich, G.S. Pinkus, et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nature Medicine, 8(1): pp. 68�74, 2002 - Huber et al., Variance stabilization applied to microarray data calibration and to the quantification of differential expression, Bioinformatics (2002) 18 Suppl.1 96-104 |
|
|
Shin et al. (2007) T-Cell Lymphoma data set |
[X] |
|
Short description: Analysis of cutaneous T-Cell lymphoma (CTCL) samples from lesional skin biopsies. Samples are divided in lower-stage (stages IA and IB, 0) and higher-stage (stages IIB and III) CTCL. Sample types: lower_stage, higher_stage No. of genes: 2922 (pre-processed) No. of samples: 63 (class 1: 20, class 0: 43) Normalization: VSN (Huber et al., 2002) References: - J. Shin, S. Monti, D. J. Aires, M. Duvic, T. Golub, D. A. Jones and T. S. Kuppe, Lesional gene expression profiling in cutaneous T-cell lymphoma reveals natural clusters associated with disease outcome. Blood, 110(8): pp. 3015, 2007 - Huber et al., Variance stabilization applied to microarray data calibration and to the quantification of differential expression, Bioinformatics (2002) 18 Suppl.1 96-104 |
|
|
Armstrong et al. (2002) Leukemia data set |
[X] |
|
Short description: Comparison of three classes of Leukemia samples: Acute lymphoblastic leukemia (ALL, 0), acute myelogenous leukemia (AML, 1) and ALL with mixed-lineage leukemia gene translocation (MLL, 3). Sample types: ALL, AML, MLL No. of genes: 8560 (pre-processed) No. of samples: 72 (class 0: 24, class 1: 28, class 2: 20) Normalization: VSN (Huber et al., 2002) References: - S.A. Armstrong, J.E Staunton, L.B. Silverman, R. Pieters, M.L. den Boer, M.D. Minden, S.E. Sallan, E.S. Lander, T.R. Golub, S.J. Korsmeyer; MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nature Genetics, 30(1): pp. 41�47, 2002 - Huber et al., Variance stabilization applied to microarray data calibration and to the quantification of differential expression, Bioinformatics (2002) 18 Suppl.1 96-104 |
|
|
SVM - Support Vector Machine |
[X] |
|
Short description: Support vector machines (SVMs) belong to the most popular methods in microarray sample classification. The SVM classifier differs from other learning algorithm in that it selects the separating hyperplane with the maximum distance to the closest samples (the maximum margin hyperplane). Extensions to the linear SVM like the "soft margin" and the "kernel trick" allow the classifier to deal with mislabelled samples and separate non-linear data. We use the linear kernel C-SVM from the e1071-package, which is a wrapper for the well-known LibSVM library. Parameter-optimization is performed via grid-search in a nested cross-validation routine. References: - Dimitriadou, E. and Hornik, K. and Leisch, F. and Meyer, D. and Weingessel, A. and Leisch, M.F. (2005), Misc functions of the department of statistics (e1071), TU Wien - C.-C. Chang and C.-J. Lin (2001), LIBSVM}: a library for support vector machines, https://www.csie.ntu.edu.tw/~cjlin/libsvm |
|
|
BioHEL |
[X] |
|
Short description: BioHEL (Bioinformatics-oriented hierarchical evolutionary learning; J. Bacardit, 2006) is a rule-based machine learning system using the concept of evolutionary learning within an interative rule learning (IRL) framework. The generated models consist of structured classification rule sets commonly known as "decision lists". These rule sets are built by iteratively learning new rules based on an almost standard generational Genetic Algorithm until the combination of rules covers all observations. Each time a new rule has been learned, the matching samples are removed from the search space. For a more detailed description of BioHEL, please consult the references. References: - Bacardit J, Krasnogor N: BioHEL: Bioinformatics-oriented Hierarchical Evolutionary Learning 2006. [eprints.nottingham.ac.uk] - Bacardit J, Burke E, Krasnogor N: Improving the scalability of rule-based evolutionary learning. Memetic Computing (to appear) |
|
|
PAM - Prediction Analysis for Microarrays |
[X] |
|
Short description: The Prediction Analysis for Microarrays (PAM; Tibshirani et al., 2002) method uses the nearest shrunken centroid approach to classify microarray samples. For each class the centroid is calculated and shrunken towards the overall centroid for all classes by a certain amount (depending on a user-defined threshold parameter). New samples are assigned to the group of the nearest centroid, but based on the shrunken centroids. The shrinkage reduces the effect of noisy genes and removes genes from the selection that are shrunken to zero for all classes. We use the standard implementation in the pamr-package and choose the threshold parameter automatically based on nested cross-validation. References: - Robert Tibshirani, Trevor Hastie, Balasubramanian Narasimhan, and Gilbert Chu (2002). Diagnosis of multiple cancer types by shrunken centroids of gene expression PNAS 99, p. 6567-6572 |
|
|
RF - Random Forest classifier |
[X] |
|
Short description: Breiman's random forest (RF) classifier uses an ensemble of unpruned decision trees to make predictions. Binary partition-trees are grown partially at random by selecting different random sub-samples of the training data for each tree and different random sub-samples of features (size m) at each node split. Although the single trees typically only have weak predictive power the ensemble often provides high accuracies, profiting from the trees' diversity introduced by the bootstrap sampling routine. We use the RF implementation from the "randomForest" R-package, set parameter m to the square root of the total number of predictors and use an ensemble of 500 trees. References: - Breiman, L. (2001), Random Forests, Machine Learning 45(1), p. 5-32 |
|
|
kNN - k-Nearest Neighbor classifier |
[X] |
|
Short description: The k-nearest neighbor (kNN) algorithm is one of the simplest machine learning algorithms, but often performs well in microarray sample classification tasks. Every new sample is assigned to the majority class of its k nearest neighbors in the training set (usually based on the Euclidean distance metric). By means of the parameter k the bias/variance tradeoff can be controlled. Since kNN does not require a training phase, the algorithm has low runtime requirements even for high-dimensional data sets. We determine the parameter k automatically using nested cross-validation. |
|
|
Help |
[X] |
Close |
|
|
ENSEMBLE |
[X] |
|
Short description: The ensemble predictor combines the SVM, PAM, RF and kNN algorithms together to obtain a more robust sample classification. Samples are assigned to the class that receives the majority of votes across the algorithms. For each of the used methods the parameter selection is performed automatically (see the descriptions of the corresponding algorithms). |
|
|
Terms and Conditions |
[X] |
Close |
|
|
Arraymining.net - Newsletter |
[X] |
|
Stay informed about updates and new features on our website by joining our newsletter. Your email address remains strictly confidential and will only be used to inform you about major updates of our web-service (<= 1 email per month). You can unsubscribe at any time by clicking on the unsubscribe link at the bottom of our e-mails. |
|
|
Arraymining.net - Newsletter |
[X] |
|
|
|

Class Assignment Analysis (Supervised Learning)
The module below allows you to perform a supervised analysis for a pre-normalized input matrix.
To obtain instructions click help.