ArrayMining - Online Microarray Data Mining
Ensemble and Consensus Analysis Methods for Gene Expression Data
 Contact Contact |
[X] |
|
The project and the website are maintained by Enrico Glaab School of Computer Science, Nottingham University, UK  webmaster@arraymining.net webmaster@arraymining.net
|
|
| Close | |
|
Help |
[X] |
Close |
|
|
Terms and Conditions |
[X] |
Close |
|
|
Empirical Bayes method |
[X] |
|
Short description: The Empirical Bayes (EB, ComBat) method by Johnson et al. combines microarray data sets by employing parametric and non-parametric empirical Bayes methods to filter out batch effects. The parameters for a location- and scale-adjustment model (which represent the additive and multiplicative batch effects) are estimated by pooling information across genes in each batch to shrink the parameter estimates towards the overall mean of the estimates across genes. This approach provides an increased robustness against outliers and can be therefore be applied to data sets with small samples sizes (for more details see the references). References: - Johnson, W.E. and Li, C. and Rabinovic, A. (2007), Adjusting batch effects in microarray expression data using empirical Bayes methods, Biostatistics, 8, p. 118 |
|
|
XPN method |
[X] |
|
Short description: The XPN cross-study normalization method by Shabalin et al. uses linked clustering of the genes and samples and a maximum likelihood approach to estimate the parameters of a block-linear error model. The underlying assumption is that the observed expression values for the genes in samples of different studies can be interpreted as scaled and shifted block means (plus additional gaussian noise), where the block means are constant across several genes and samples, and the same in each study. For more details about the method and a comparative evaluation with other approaches, please refer to the original publication by Shabalin et al. (see references) References: - Shabalin , A. A., Tjelmeland, H., Fan, C., Perou, C. M., and Nobe, A. B. (2008). Merging two gene-expression studies via cross-platform normalization. Bioinformatics, 24, p. 1154-1160 |
|
|
Median Rank Score Normalization (MNORM) |
[X] |
|
Short description: The Median Rank Score Normalization method (MNORM) by Warnat et al. is a very fast and simple approach which replaces the expression values for a single study by median expression values of genes from another study (the "reference" study). First, the relative rank of the expression values to be replaced in the non- reference set is computed. In the second step, these expression values are replaced by the median-expression values from the reference-study whose positions in a sorted vector correspond to the ranks in the non-reference set. More details can be found in the paper by Warnat et al. References: - Warnat, P. and Eils, R. and Brors, B. (2005), Cross-platform analysis of cancer microarray data improves gene expression based classification of phenotypes, BMC Bioinformatics, 6, p. 265 |
|
|
Quantile discretization normalization |
[X] |
|
Short description: The quantile discretization method for cross-study normalization by Warnat et al. combines microarray data sets by first discretizing them. The expression values for each study are assigned to 8 different bins given by the the quantiles of the array expression values as cut-points. To obtain the final discretized values, the two central bins are merged and all expression values are replaced by the number of the bin they were assigned to. This simple approach can result in a considerable loss of information, but can be useful when the data is very noisy or the user is intending to apply statistical analysis methods which require discretized values as input. References: - Warnat, P. and Eils, R. and Brors, B. (2005), Cross-platform analysis of cancer microarray data improves gene expression based classification of phenotypes, BMC Bioinformatics, 6, p. 265 |
|
|
NorDi discretization normalization |
[X] |
|
Short description: The NorDi method by Martinez et al. is a discretization approach, which can also be used as a simply means to combine data from different studies (similar to the QDISC method). NorDi (= Normalized Discretization) improves the normality of the gene expression values (on logarithmic scale) by removing outliers using the Grubbs method and the Jaque–Bera normality test. The method then determines significantly over- or under-expressed genes by computing z-scores (Yang et al., 2002) for each gene. The original gene expression values are then discretized based on z-score thresholds using three intervals ("under-expressed", "normal", "over-expressed"). References: - Martinez, R. and Pasquier, N. and Pasquier, C. (2008), GenMiner: mining non-redundant association rules from integrated gene expression data and annotations, Bioinformatics, 24, p. 2643 |
|

Cross-Study Normalization
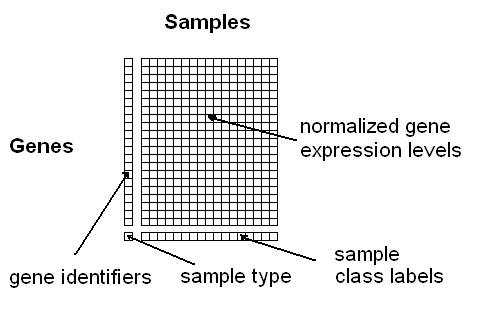
This module provides methods to combine microarray data from different studies and platforms together into a single data set. The input data sets need to be derived from the same tissue type under comparable biological conditions and the genetic probes must overlap. To obtain instructions click help.